

With the explosive growth of computer systems in the life sciences industry and automation playing an increasing role in development, the need for securing electronic records is more crucial than ever. 21 CFR Part 11 works with predicate rules (requirements set forth by any FDA regulation) that require companies to maintain records as part of their compliance.
How do you become FDA 21 CFR Part 11 Compliant?
To be fully compliant with 21 CFR Part 11, your entire team needs to understand how to treat documents and signatures. The following is a checklist to identify current areas of risk and adhere to key components of compliance.
Highlights of FDA 21 CFR Part 11
Related Read: End to End Development of a Wearable Gastro-recorder
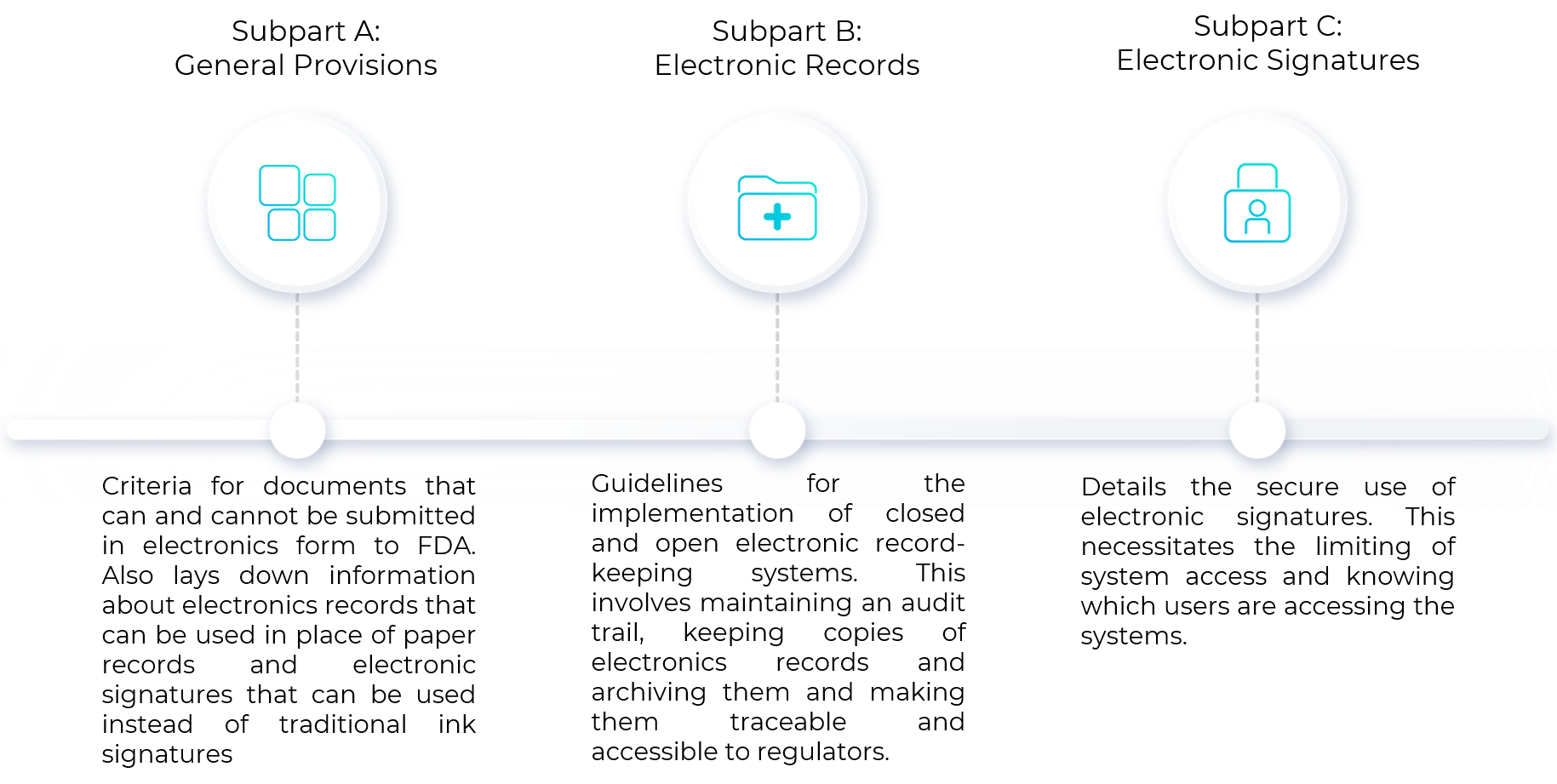
Sub-part A: General Provisions
This sub-part/section encompasses the scope of compliance to 21 CFR Part 11 and relevant definitions. It deals with general information about the qualification criteria for FDA to accept the documents that can be submitted in electronic form. This section also provides information about the documents that cannot be submitted to FDA in electronic form, superseding criteria to meet other agency regulations and criteria for documents that need to be maintained but not submitted to FDA. Electronics records can be used in place of paper records and electronic signatures can be used in lieu of traditional ink signatures, in whole or in part, provided the below requirements are met:
- Records in electronic form are created, modified, maintained, archived, retrieved, or transmitted according to the requirements set forth by Part B of 21 CFR Part 11.
- Electronic records are created on or after August 20, 1997.
- Electronic signatures are equivalent to handwritten signatures according to the requirements set forth by Part C of 21 CFR Part 11
- Computer systems (including hardware and software), controls, and attendant documentation are readily available for FDA inspection if necessary.
Sub-part B: Electronic records
This section deals with guidelines for the implementation of closed and open electronic record-keeping systems. Closed systems refer to those in which access is controlled by individuals responsible for content generation. Open systems refer to those in which access is controlled by a third-party and not by individuals responsible for content generation. At a high level, this involves maintaining an audit trail, keeping copies of electronics records and archiving them and making them traceable and accessible to regulators. Successful implementation of the following requirements ensures compliance to Sub-part B of 21 CFR Part 11:
- Procedures and controls to ensure the authenticity, integrity, and confidentiality of electronic records in closed and open systems. Integrity and accuracy of the records can be ensured by validating the source of data input, and by rejecting the invalid and incomplete data.
- Limiting access to the system to authorized individuals only. This can be achieved by performing authentication checks and authorization checks to ensure that only authorized individuals can use the appropriate features of the system, electronically sign a record or alter a record.
- Accurate and complete generation of copies of records by the system, in both human-readable and electronic form, for inspection, review, and copying by the FDA.
- Protection, retention and ready retrievability of electronic records throughout the record retention period.
- Audit trail that captures and stores the date & time of entries, actions that create, modify, or delete electronic records. Ensure audit trail includes the User ID, original and new values of altered / new records as appropriate. Also ensure that the audit trail does not overwrite log files throughout the retention period.
- Establishment and adherence to an official policy to prevent signature falsification.
- Maintenance of documentation (system operation and maintenance related documents) using version control system
- Enforcement of permitted sequence of steps and events as appropriate. This can be done by implementing proper work flow.
- Providing appropriate training to individuals responsible for developing and maintaining the electronic records
- Protection of electronic records and signatures during transit (Applicable for ‘Open’ systems). This can be achieved by encrypting the data and by using digital signatures/certificates.
- Signed electronic records contain the printed name of the signer, the date and time of signature and the meaning of the signature (such as approval, review, etc.)
- Binding of electronic signatures to their respective electronic records to protect against falsification.
Sub-part C: Electronic Signatures
Another crucial aspect of adhering to 21 CFR Part 11 is the secure use of electronic signatures. This necessitates the limiting of system access and knowing which users are accessing the database. Successful implementation of the following requirements ensures compliance to Sub-part C of 21 CFR Part 11:
- Electronic signature is unique to an individual, never reused or reassigned to another user
- Procedure and controls to verify the identity of an individual prior to allocating an electronic signature
- Ensuring that non bio-metric electronic signature is made up of at least two components, for instance, an identification code and a password
- Ensuring bio-metric electronic signatures can be used only by their genuine owner
- Password is requested each time, when electronic signatures are used several times during a session. When electronic signatures are used in separate sessions, both components of the electronic signature are required for each signing
- Attempted falsification of an electronic signature would require the collusion of at least two individuals. This can be enforced by ensuring database access is restricted to only admin user, in a live system.
- System maintains the uniqueness of each identification code and password combination, such that no two individuals have the same combination of identification code and password
- Ensuring periodic expiry and revision of passwords. This can be done by ensuring that passwords automatically expire after a certain number of days and that the same password cannot be used again when change password operation is performed.
- Recall of identification codes and passwords in cases where an individual leaves the organization or is transferred to another location
- Electronic disabling of identification code or password if they are potentially lost or compromised
- System can detect and report attempts at unauthorized use
- Established procedures for loss management to combat cases where a device is lost or stolen
- Procedures and controls exist for the issuance of temporary and permanent replacements of electronic signatures
- Initial and periodic testing of systems or devices that issue / generate identification codes to ensure they function properly
Related Read: Embedded Control Software Suite for Pharmaceutical Instruments
What to Watch Out for
FDA 21 CFR Part 11 has a range of features that are required when implementing a computer system to manage electronic records and processes. Prominent among these include electronic signatures, audit trails, security and data integrity. There are also Standard Operating Procedures (SOPs) to govern and describe how these features are enforced. Additionally, there must also be controls to allow users to identify when the system does not function as per its intended use. Compliance to FDA 21 CFR Part 11 helps the Life Science community to streamline business processes and reduce turnaround time, human errors and costs.